
Cloning & Synthetic Biology
Overview of molecular cloning |
Molecular cloning refers to the process by which recombinant DNA molecules are produced and transformed into a host organism, where they are replicated. A molecular cloning reaction is typically comprised of the following two components:
- The DNA fragment of interest to be replicated
- A vector/plasmid backbone that contains all of the components for replication in the host
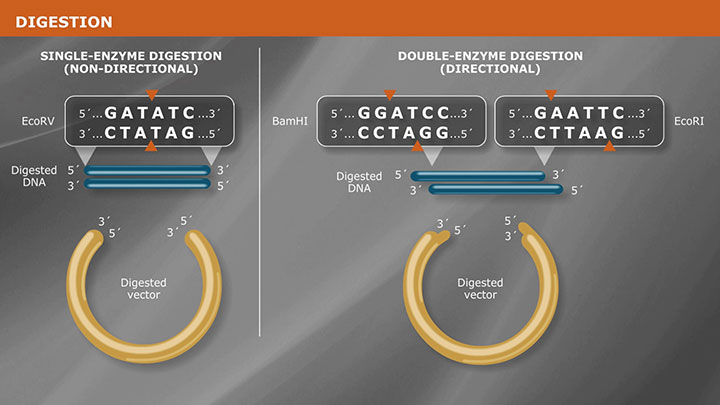
DNA of interest, such as a gene, regulatory element(s), or operon, etc., is prepared for cloning by excising it out of the source DNA using restriction enzymes, copying it using the Polymerase Chain Reaction (PCR), or assembling it from individual oligonucleotides. At the same time, a plasmid vector is prepared in linear form using restriction enzymes or PCR. The plasmid is a small, circular piece of DNA that is replicated within the host, and exists separately from the host’s chromosomal or genomic DNA. By physically joining the DNA of interest to the plasmid vector through phosphodiester bonds, the DNA of interest becomes part of the new recombinant plasmid and is replicated by the host.
Plasmid vectors allow the DNA of interest to be copied in large amounts and, often, provide the necessary control elements to be used to direct transcription and translation of the cloned DNA. As such, they have become the workhorse for many molecular methods, such as protein expression, gene expression studies, and functional analysis of biomolecules. During the cloning process, the ends of the DNA of interest and the vector have to be modified to make them compatible for joining through the action of a DNA ligase, recombinase, or in vivo DNA repair mechanism. These steps typically utilize enzymes, such as nucleases, phosphatases, kinases and/or ligases. Many cloning methodologies and, more recently, kits have been developed to simplify and standardize these processes, including those for seamless cloning and DNA Assembly.
History of molecular cloning |
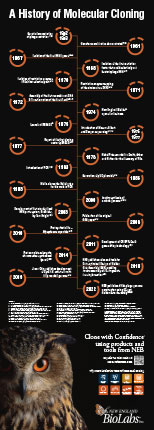 In 1952 with the genetic demonstration of phage restriction, the existence of mechanisms that protect bacteria from viral infections was revealed. This led to the isolation of the first restriction factor that could selectively cut bacteriophage DNA, in 1968. Over the following decades, scientists made significant advancements in understanding DNA replication and DNA modifying enzymes, leading to the development of various cloning techniques. Major milestones that revolutionized the field of molecular cloning during this time include the introduction of PCR in 1983, Golden Gate Assembly in 2008, and CRISPR-Cas9 gene editing in 2011.
In 1952 with the genetic demonstration of phage restriction, the existence of mechanisms that protect bacteria from viral infections was revealed. This led to the isolation of the first restriction factor that could selectively cut bacteriophage DNA, in 1968. Over the following decades, scientists made significant advancements in understanding DNA replication and DNA modifying enzymes, leading to the development of various cloning techniques. Major milestones that revolutionized the field of molecular cloning during this time include the introduction of PCR in 1983, Golden Gate Assembly in 2008, and CRISPR-Cas9 gene editing in 2011.
This poster highlights a selection of some of the most significant discoveries, advancements, and achievements in the field of molecular cloning.
View PDF Request a copy
Types of molecular cloning |
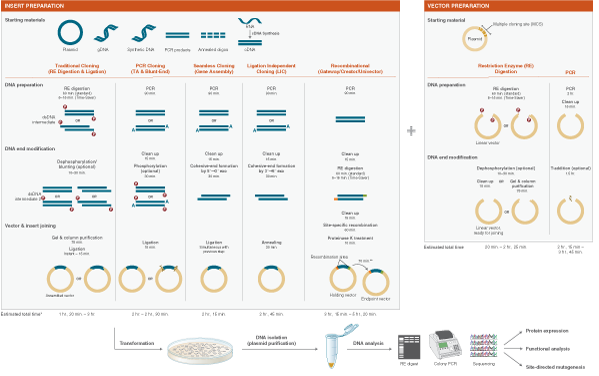
Molecular cloning techniques are diverse and new methods are continuously being developed. Most share some commonalities such as the traditional use of restriction endonucleases to cut DNA, ligases to join DNA, and DNA polymerases to copy DNA. More recent cloning methodologies utilize other properties of DNA polymerases, such as exonuclease activity, along with homologous regions of DNA that join seamlessly together.
Learn more about the various types of molecular cloning:
Synthetic biology & DNA assembly |
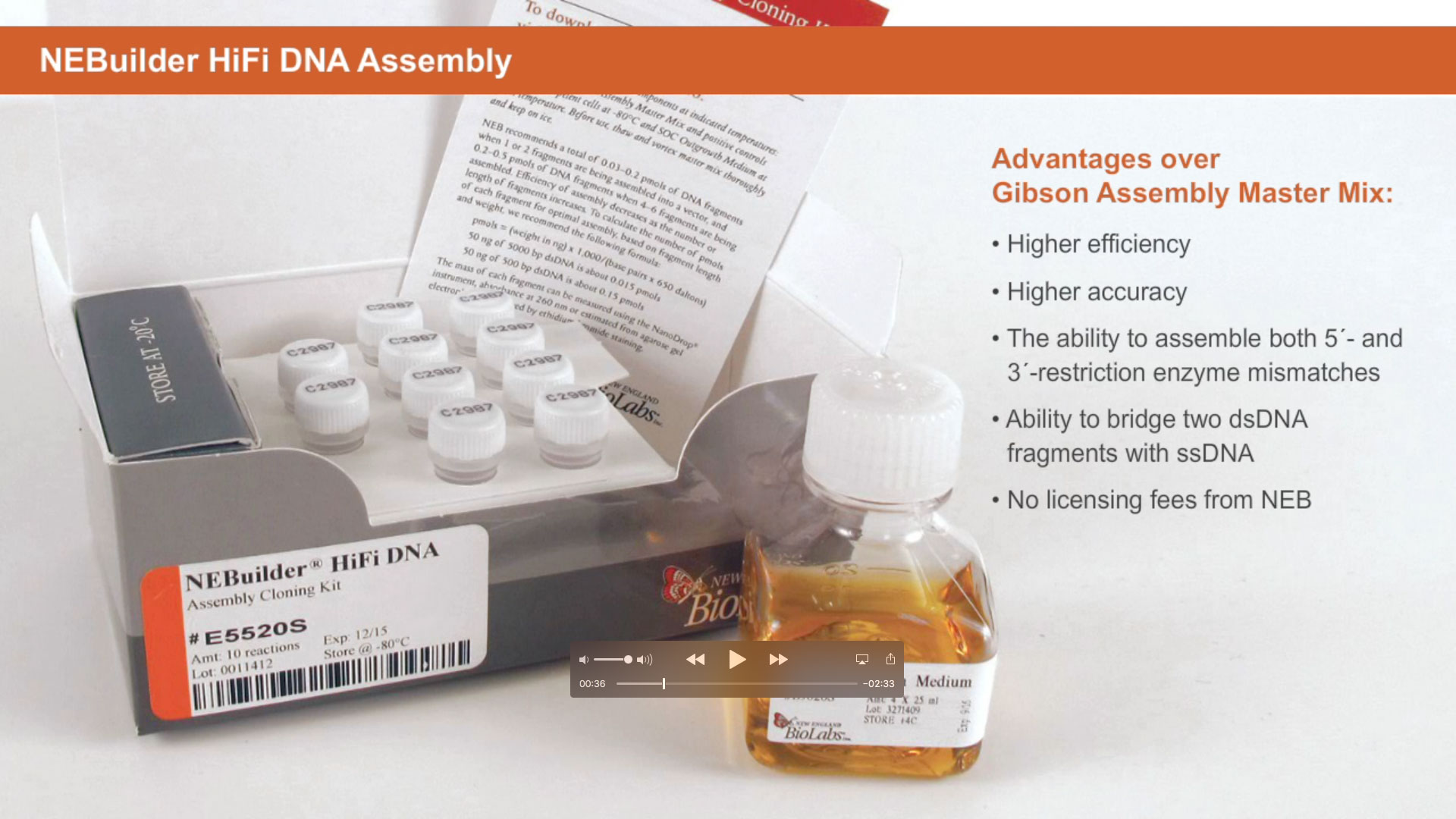
Synthetic Biology is a more recent expansion of the biotechnology field, in which genes and proteins are viewed as parts or devices, with the goal of re-designing and/or assembling these parts in novel ways to create a new and useful functionality. Recent advances in biofuels generation, production of biochemicals, and understanding the minimal genome all benefit from synthetic biological approaches. Often these projects rely on the ordered assembly of multiple DNA sequences to create large, artificial DNA structures. To this end, methods have evolved to simplify this process. NEBuilder HiFi DNA Assembly, Gibson Assembly®, and NEBridge Golden Gate Assembly can be used to create many functional DNA structures, from a simple joining of two metabolic genes, all the way up to the creation of an artificial genome.
To help select the best DNA assembly method for your needs, please visit our Synthetic Biology/DNA Assembly Selection Chart.
 Learn more about NEBuilder HiFi DNA Assembly at NEBuilderHiFi.com
Learn more about NEBuilder HiFi DNA Assembly at NEBuilderHiFi.com Learn more about NEBridge Golden Gate Assembly at neb.com/GoldenGate
Learn more about NEBridge Golden Gate Assembly at neb.com/GoldenGate
Choose Type:
includes these areas of focus:
- DNA Analysis
- Colony PCR
- DNA Sequencing
- Restriction Enzyme Digestion
- DNA Assembly and Cloning
- NEBuilder® HiFi DNA Assembly
- NEBridge® Golden Gate Assembly
- Gibson Assembly®
- BioBrick® Assembly
- DNA End Modification
- Dephosphorylation
- Phosphorylation (Kinase)
- A-tailing
- Blunting
- DNA Ligation
- Non-Cloning Ligation
- Cloning Ligation
- DNA Preparation
- Reverse Transcription (cDNA Synthesis)
- Restriction Enzyme Digestion
- PCR
- Fast Cloning: Accelerate your cloning workflows with reagents from NEB
- High-throughput cloning and automation solutions
- Nucleic Acid Purification
- RNA Cloning
- Site Directed Mutagenesis
- Transformation
- USER® Cloning
- Applications of USER® and Thermolabile USER II Enzymes
- Insert Screening Protocols for NEB PCR Cloning Kit
- Ligation Protocol for NEB PCR Cloning Kit
- Plating Protocol for NEB PCR Cloning Kit
- Transformation Protocol for NEB PCR Cloning Kit
- In vitro digestion of DNA with EnGen® Spy Cas9 HF1 (NEB #M0667)
- Determining Genome Targeting Efficiency using T7 Endonuclease I
- NEBuilder HiFi DNA Assembly Reaction Protocol
- NEBuilder HiFi DNA Assembly Transformation Protocol
- NEBuilder® HiFi DNA Assembly Chemical Transformation Protocol (E2621, E5520, E2623)
- Using recombinant Cas9 nuclease to assess locus modification in genome editing experiments (#M0386)
- Transfection of Cas9 RNP (ribonucleoprotein) into adherent cells using the Lipofectamine® RNAiMAX
- RNA Synthesis of Cloned Insert Transcripts
- Genomic DNA Cleanup Protocol
- In vitro digestion of plasmid DNA with EnGen SpRY Cas9 (NEB #M0669)
- In vitro digestion of DNA with Cas9 Nuclease, S. pyogenes (M0386)
- NEBuilder® HiFi DNA Assembly Electrocompetent Transformation Protocol
- Transfection of EnGen® Spy Cas9 HF1 (NEB #M0667) into adherent cells using the Lipofectamine® RNAiMAX System
- Improved methods for site-directed mutagenesis using Gibson Assembly® Master Mix
- Robust Colony PCR from Multiple E. coli Strains using OneTaq® Quick-Load® Master Mixes
- Improved methods for site-directed mutagenesis using NEBuilder® HiFi DNA Assembly Master Mix
- Improved method for assembly of linear yeast expression cassettes using NEBuilder® HiFi DNA Assembly Master Mix
- Enhancing Transformation Efficiency
- Protocol for using Recombinant Cas9 Nuclease to Assess Locus Modification in Genome Editing Experiments
- Quick Ligation Kit
- Nanoliter Scale DNA Assembly Utilizing the NEBuilder HiFi Cloning Kit with the Labcyte Echo 525 Liquid Handler
- Fast & efficient isolation of phage genomic DNA using the Monarch HMW DNA Extraction Kits
- Accelerating DNA Construction to Protein Expression A Rapid 1-Day Workflow Using NEBridge Golden Gate Assembly
- A faster workflow for the assessment of genomic loci in mice using a novel HMW DNA extraction technology upstream of Cas9 targeted sequencing
-
Restriction Endonucleases: Molecular Cloning and Beyond
-
A Modern Day Gene Genie Sir Richard Roberts on Rebase
-
How Gibson Assembly® is Changing Synthetic Biology
Understand how Gibson Assembly ® works and its impact in accelerating the progress of synthetic biology.
- Competent Cell Brochure
- DNA Assembly & Synthetic Biology Brochure
- Molecular Cloning Technical Guide
- NEBuilder HiFi DNA Assembly Bifold
- PCR Reagents Brochure
- NEBcloner®
- NEBioCalculator®
- PCR Selector
- Cleavage Of Supercoiled DNA
- Cloning Plasmids and DNAs
- Compatible Cohesive Ends and Generation of New Restriction Sites
- Competent Cell Selection Guide
- DNA Ligase Selection Chart
- DNA Markers & Ladders Selection Tool
- Dam-Dcm and CpG Methylation
- Frequencies of Restriction Sites
- Recleavable Blunt Ends
- Recleavable Filled-in 5' Overhangs
- Synthetic Biology/DNA Assembly Selection Chart
- Why Choose Recombinant Enzymes?
- PCR Troubleshooting Guide
- Troubleshooting Guide for Cloning
- Troubleshooting Tips for Ligation Reactions
- Activity at 37°C for Restriction Enzymes with Alternate Incubation Temperatures
- Chemical Transformation Tips
- Cleavage Close to the End of DNA Fragments
- Digestion of Agarose-Embedded DNA: Info for Specific Enzymes
- Double Digests
- Electroporation Tips
- Getting Started with Molecular Cloning: Simple Tips to Improve your Cloning Efficiency
- Optimizing Restriction Endonuclease Reactions
- Restriction Endonucleases - Survival in a Reaction
- Site Preferences
- Star Activity
- Traditional Cloning Quick Guide
- Prediction of Golden Gate Assembly GGA Using a Comprehensive Analysis of T4 DNA Ligase End-Joining Fidelity and Bias (2018)
Feature Articles
Brochures
Web Tools
Selection Tools
Troubleshooting Guides
Usage Guidelines
Posters
- Anton, B.P., Morgan, R.D., Ezraty, B., Manta, B., Barras, F., Berkmen, M. (2019) Complete genome sequence of Escherichia coli BE104, an MC4100 drivative lacking the methionine reductive pathway Microbiol Resour Announc; 8 (29), e00721-19. PubMedID: 31296691, DOI: 10.1128/MRA.00721-19
- Gehring, A.M., Zatopek, K.M., Burkhart, B.W., Potapov, V., Santangelo, T.J., Gardner, A.F (2019) Biochemical reconstitution and genetic characterization of the major oxidative damage base excision DNA repair pathway in Thermococcus kodakarensis DNA Repair (Amst); PubMedID: 31841800, DOI: 10.1016/j.dnarep.2019.102767
- Potapov, V., Ong, J.L., Kucera, R.B., Langhorst, B.W., Bilotti, K., Pryor, J.M., Cantor, E.J., Canton, B., Knight, T.F., Evans, T.C., Lohman, G.J.S. (2018) Comprehensive profiling of four base overhang ligation fidelity by T4 DNA ligase and application to DNA assembly ACS Synth Biol; 7 (11), PubMedID: 30335370, DOI: 10.1021/acssynbio.8b00333
- Ke, Na; Berkmen, Mehmet; Ren, Guoping; (2017) A water-soluble DsbB variant that catalyzes disulfide-bond formation in vivo Nat Chem Biol; 13, 1022-1028. PubMedID: 28628094, DOI: 10.1038/nchembio.2409
- Shah, S., Sanchez, J., Stewart, A., et al. (2015) Probing the Run-On Oligomer of Activated SgrAI Bound to DNA PLoS One; 10(4), PubMedID: 25880668, DOI: 10.1371/journal.pone.0124783.
- Roberts, R.J., Vincze, T., Posfai, J., Macelis, D. (2015) REBASE - A database for DNA restriction and modification: enzymes, genes and genomes Nucleic Acids Res; 43, D298-D299. PubMedID: 25378308
- Roberts, R.J., Vincze, T., Posfai, J., Macelis, D. (2014) REBASE - A database for DNA restriction and modification: enzymes, genes and genomes Nucleic Acids Res;
- Mauris, J.and Evans, T.C., Jr. (2010) A human PMS2 homologue from Aquifex aeolicus stimulates an ATP-dependent DNA helicase. J Biol Chem; 285(15), 11087-11092. PubMedID: 20129926
Products and content are covered by one or more patents, trademarks and/or copyrights owned or controlled by New England Biolabs, Inc (NEB). The use of trademark symbols does not necessarily indicate that the name is trademarked in the country where it is being read; it indicates where the content was originally developed. The use of this product may require the buyer to obtain additional third-party intellectual property rights for certain applications. For more information, please email busdev@neb.com.
This product is intended for research purposes only. This product is not intended to be used for therapeutic or diagnostic purposes in humans or animals.
-
Overview of Traditional Cloning
-
Introduction to NEBUILDER HIFI DNA ASSEMBLY®
-
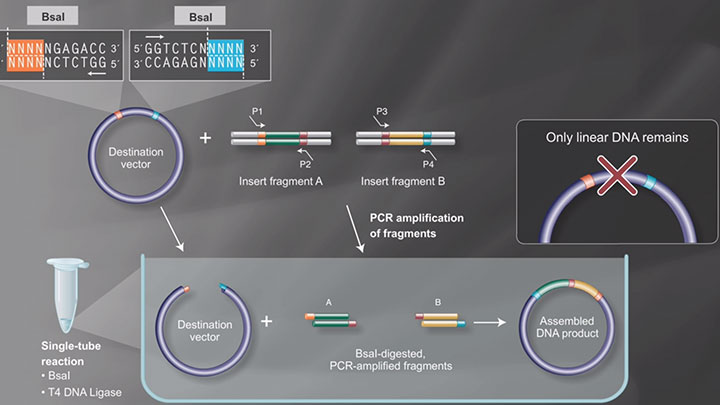
Golden Gate Assembly Workflow
-
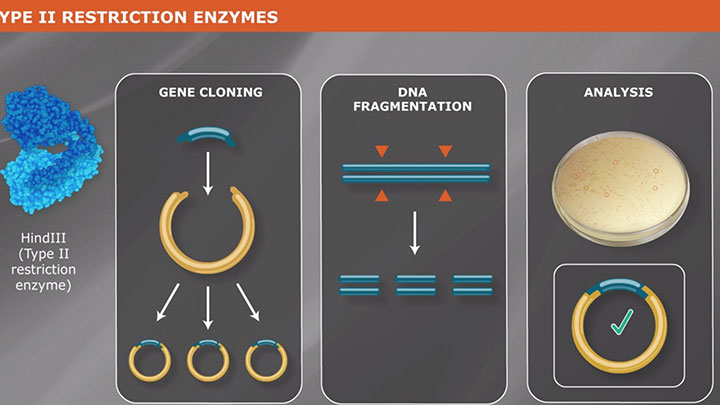
What is a Type II Restriction Enzyme?
-
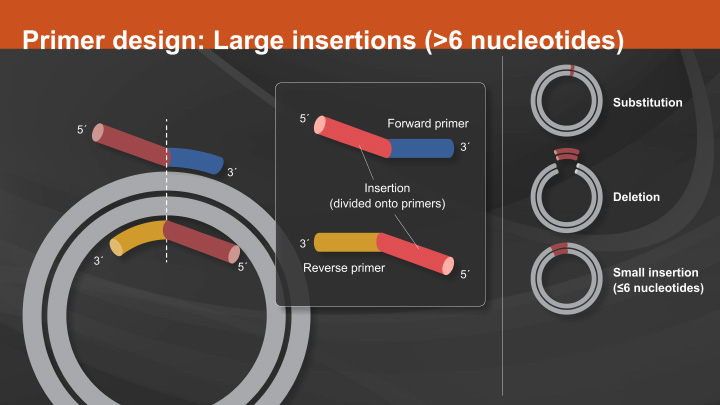
Overview of the Q5® Site-Directed Mutagenesis Kit
-

DNA Blunting Tutorial
-

The Mechanism of Transformation with Competent Cells
-
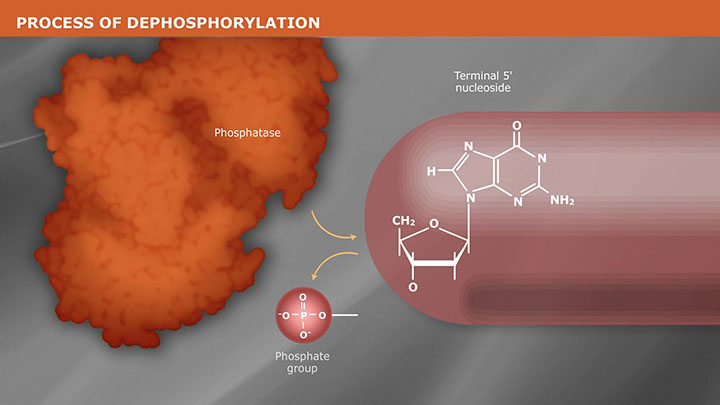
The Mechanism of Dephosphorylation
-
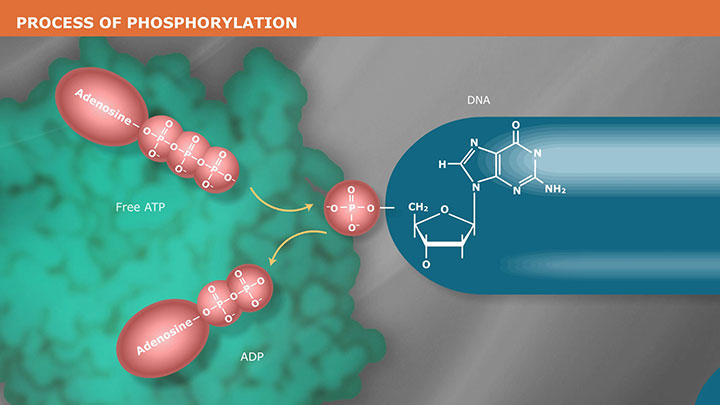
The Mechanism of DNA Phosphorylation
-
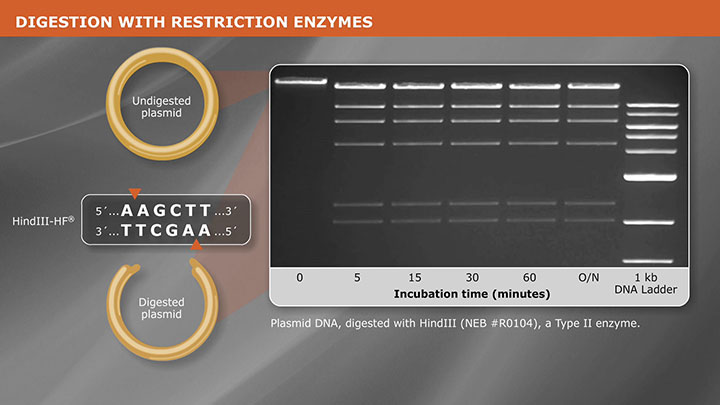
Cloning With Restriction Enzymes
Need more help selecting
a product for DNA assembly?
![]()
View our Synthetic Biology/DNA Assembly Selection Chart for more details.
Which molecular cloning
technique is best for you?
![]()
For the new cloner, NEB suggests choosing one of three cloning methods. Find the method that works for your application.
Download the Molecular Cloning Technical Guide
![]()
Download this technical guide for help with product selection, protocols, tips for optimization and troubleshooting.
Download Tech Guide

